About Heligeom
Heligeom aims at characterizing, manipulating and assembling structural units with a screw organization,
in which the structural units may be individual proteins or protein hetero-multimers.
Heligeom relies on structures of monomer-monomer interfaces both for defining the transformation and for constructing representative filaments.
Screw & Helical Parameters
Heligeom is based on a representation of an arbitrary spatial displacement of a rigid body in terms of a screw transformation.
A screw motion is defined by the six degrees of freedom associated with a rotation in 3D around a specified axis and a translation along this axis.
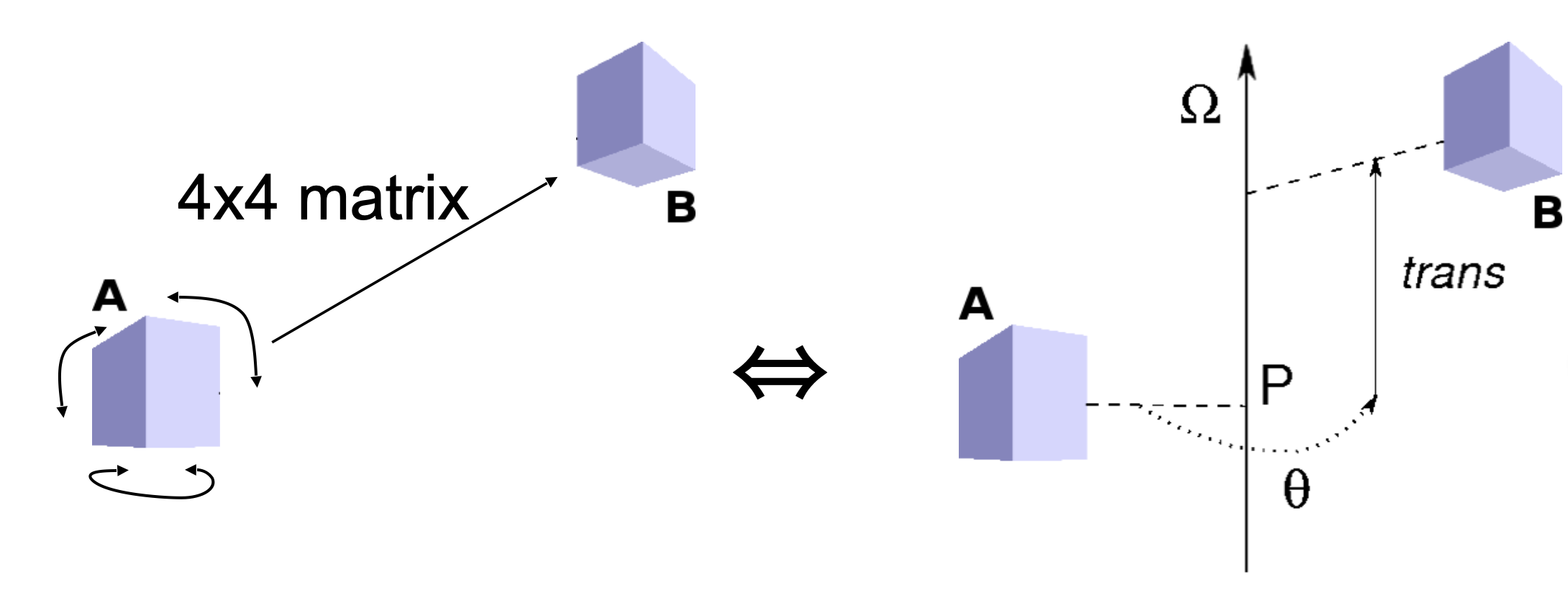
The Heligeom webserver is based on the PTools library,
in which screw motions are calculated and stored in a compact implementation from the 4x4 rotation-translation matrix provided by superposition of the input protein monomers. The user may also specify a quasi-rigid core region of the input proteins for defining the screw transformation.
The screw transformation is defined by:
- the position P and direction Ω of a screw axis
- a rotation θ around this axis
- a translation trans parallel to the axis
Parameters describing the helix or ring shape, i.e. the pitch, the number of monomers per turn nb and the direction of rotation dir, are derived from the screw parameters as follows:
- The pitch is the distance of one helical turn measured along the helix axis.
- The monomer per turn is the number of monomers needed for exactly one turn of the helix.
- The Handedness is the direction of rotation around the axis.
- The 2 distances (IntR and ExtR) are the minimal and maximal distances between the screw axis and the atoms of the oligomeric filament.

Comparison of interfaces
If two interfaces are provided, the server provides both sets of helical parameters and additionally computes the fNAT : the fraction of residue-residue contacts shared in the two interfaces, which is a measure of interface similarity commonly used to evaluate protein-protein docking predictions (see the home page for an example.)
Construction of one oligomer
Based on the Screw geometry, the Heligeom web server can construct structures of desired length (the number of assembled monomers is limited to 100), which can be downloaded as .pdb or .mmcif files.
For convenience, the server can be requested to align the helix axis of the generated complex with the Z-axis.
In case the helical analysis has been restricted to a core protein region (see above), the whole protein may nevertheless be used for construction, in which case
all regions will undergo the screw transformation defined by the core region.
Targeted adjustment (i.e Flattening)
Heligeom can be used to adjust an experimental or a predicted helical assembly having a small pitch and a number of monomers per turn close to an integer value N
to attain a ring structure, with zero pitch and precisely N monomers per turn, using a method described in [1].
In short, after calculating the screw parameters for two interacting subunits, the adjustment process consists
in slightly re-positioning and re-orienting the first subunit with respect to the screw axis, which is kept fixed, in such a way that
an acceptable interaction energy is obtained between subunit 1 and its image
obtained via rotation by 2π/N in the plane perpendicular to the axis.
Initial radial positioning of the first subunit center of mass with respect to the axis is evaluated using simple geometrical considerations.
Subsequently, an exploration is carried out using concerted rigid-body moves consisting of radial translations of the interacting subunits and rotations around their centers of mass,
which are accepted or rejected based on a Metropolis Monte Carlo criterion.
The process employs a reduced representation of the proteins [2] in order to decrease the sensitivity to side chain conformational variability at the interface.
References
- Boyer B, Ezelin J, Poulain P, Saladin A, Zacharias M, Robert C H, and Prévost C (2015)
An Integrative Approach to the Study of Filamentous Oligomeric Assemblies, with Application to RecA. PLOS ONE 10(3): e0116414.
https://doi.org/10.1371/journal.pone.0116414
- M. Zacharias (2003) Protein-protein docking with a reduced protein modelaccounting for side-chain flexibility, Protein Sci 12 (6) 1271–1282 https://doi.org/10.1110/ps.0239303